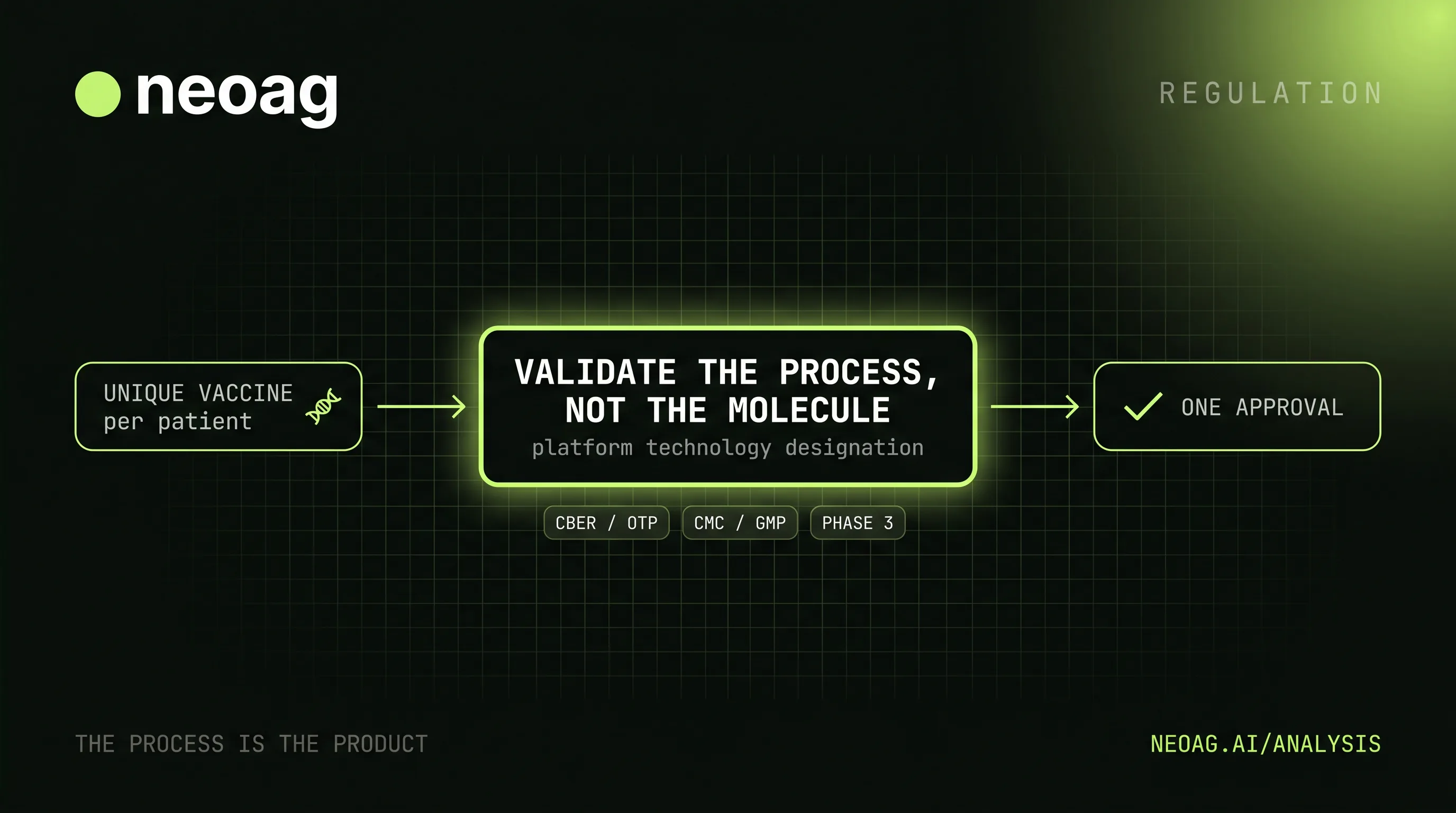
A personalized cancer vaccine is, by construction, a different medicine for every patient. Each one is built from the specific somatic mutations in a single tumor — Moderna and Merck's intismeran autogene (mRNA-4157 / V940) encodes up to 34 neoantigens drawn from one patient's mutational signature, so no two doses are the same molecule. That collides head-on with a regulatory system designed around the opposite assumption: a drug is one defined, reproducible product, characterized batch after identical batch. If the product is different every time, what exactly is the FDA approving?
This is the question the field's commercial future turns on, and it is usually filed under the label "N-of-1." That label hides a useful distinction — and the regulators' answer is more settled, and more pragmatic, than the headlines suggest. This piece is the regulatory companion to the field's computational blueprint: not how the vaccine is designed, but how a medicine that is unique to each patient is allowed to reach them.
The answer in one minute
FDA does not need every patient's neoantigen sequence to become a separate full drug application. The agency can regulate the repeatable platform: tumor sequencing, variant calling, neoantigen selection, synthesis, formulation, release testing, stability, chain of identity, and clinical-use rules. The individualized sequence changes; the controlled process and acceptance criteria are what must be validated.
That makes CMC — chemistry, manufacturing, and controls — the bottleneck. A sponsor has to show that thousands of bespoke lots can be made on time, tested consistently, and compared across process changes. Clinical efficacy still has to come from ordinary multi-patient trials. The product is individualized, but the evidence package is not a one-patient anecdote.
"N-of-1" — a product, not a trial
The phrase N-of-1 gets used two ways, and conflating them causes most of the confusion. A true N-of-1 trial is a formal study design: a single patient, cycled through treatment and control in a multiple-crossover experiment, where N=1 is literally the sample size and the unit of analysis. That is not what cancer-vaccine companies run. They run conventional, multi-patient, randomized cohort trials — it is the product that is individualized, not the study design.
So the precise statement is that these programs test N-of-1 products inside ordinary clinical trials. Every enrolled patient gets a differently sequenced vaccine, but the trial randomizes a cohort against a control arm and pools the outcomes, exactly as any phase III would. When a regulator evaluates the result, the intervention being judged is the platform and its process — not any one patient's molecule. Read "N-of-1 cancer vaccine" as a statement about manufacturing, not about statistics.
- Single-patient (N-of-1) trials — what the term actually means — The formal crossover design, distinct from an individualized product in a cohort trial
- Individualized mRNA neoantigen vaccine, randomized cohort trial (Nature, 2025) — Personalized product, conventional randomized study design
FDA's answer: regulate the process, not the molecule
The reconciliation is an idea industry shorthand calls "the process is the product": when the final molecule can't be held constant, you validate and lock the manufacturing process and release specifications instead, and treat that validated process as the thing being approved. The FDA already runs autologous cell therapies — CAR-T, sipuleucel-T — this way, where every dose is patient-specific but the starting materials and processing steps are fixed and audited. (The slogan is industry usage, not an FDA term of art; the underlying doctrine is FDA's comparability framework for biologics.)
FDA has since given this a formal vehicle. Its draft Platform Technology Designation Program, published in May 2024, defines a platform as a "well-understood and reproducible" technology that can be adapted to more than one product through a standardized manufacturing process — and explicitly names mRNA/lipid-nanoparticle systems as a candidate. Once a platform is designated, a sponsor can carry prior manufacturing and stability data forward as "prior knowledge" to support the next product built on it, rather than re-proving the process from scratch each time. That is precisely the lever an individualized-vaccine maker needs. The program is still draft guidance, and untested for a marketed individualized cancer vaccine.
- FDA — Platform Technology Designation Program (draft guidance, May 29 2024) — The formal "hold the process constant" pathway; names mRNA/LNP
- FDA — Demonstration of Comparability of Human Biological Products — The comparability doctrine under the "process is the product" idea
The N-of-1 precedent the FDA already set — milasen
The agency has done genuinely single-patient regulation before. In 2018 a splice-correcting antisense oligonucleotide, milasen, was designed, manufactured, and dosed under an individual-patient IND for one child with a unique Batten-disease mutation — from identification to first dose in about a year. That case prompted the FDA to publish a suite of draft guidances on "Individualized Antisense Oligonucleotide" drug products (2020–2022), spelling out how to run an IND for a therapy built for a single identified person.
Those guidances are the clearest evidence that the FDA is willing to bend its product-centric framework for individualized medicine — but they are only a partial template for cancer vaccines. They are scoped to ultra-rare genetic disease affecting one or two patients, and aimed at sponsor-investigators; they describe something close to a true single-patient study. Neoantigen vaccines, by contrast, run multi-patient cohort trials under a single corporate IND. The ASO work is the conceptual precedent for "individualized product, streamlined oversight," not a literal pathway a vaccine sponsor files under.
- NEJM — Patient-Customized Oligonucleotide Therapy (milasen, 2019) — The single-patient ASO that triggered FDA's individualized-therapy guidances
- FDA — IND Submissions for Individualized ASO Drug Products (Federal Register, Jan 5 2021) — The foundational draft framework for regulating a drug made for one person
Where real programs sit with the FDA
No individualized neoantigen vaccine is FDA-approved. The furthest-along program is Moderna and Merck's intismeran autogene plus pembrolizumab, which won Breakthrough Therapy designation in adjuvant melanoma in February 2023 on the strength of the phase 2b KEYNOTE-942 trial — a 44% reduction in the risk of recurrence or death (recurrence-free survival, HR 0.56), a benefit that held at five years (a 49% reduction). A designation accelerates FDA interaction; it is not an approval. The category's pivotal test is the phase 3 INTerpath-001 trial in resected high-risk melanoma, with a companion phase 3 in non-small-cell lung cancer — randomized, hard-endpoint readouts that will decide whether the platform model is real.
The other closely watched program, BioNTech and Genentech's autogene cevumeran, holds FDA orphan-drug designation in pancreatic cancer and is in randomized phase 2 (IMCODE-003); a Breakthrough or RMAT designation for it is not on the public record. The pattern across the field is consistent: the FDA hands out its expedited-development tools readily, but the bar for approval remains a randomized phase 3 showing a clinical-benefit endpoint, not a surrogate immunogenicity signal.
| Program | Sponsor | Lead indication | US regulatory status |
|---|---|---|---|
| Intismeran autogene (mRNA-4157 / V940) | Moderna / Merck | Adjuvant melanoma | Breakthrough Therapy (Feb 2023); phase 3 INTerpath-001 ongoing |
| Autogene cevumeran (BNT122) | BioNTech / Genentech | Resected pancreatic (PDAC) | Orphan Drug; randomized phase 2 (IMCODE-003) |
| Any individualized neoantigen vaccine | — | — | No FDA approval to date |
- Merck — Breakthrough Therapy designation for mRNA-4157/V940 (Feb 22 2023) — The field's lead designation, on KEYNOTE-942 data
- ClinicalTrials.gov — INTerpath-001 (NCT05933577), phase 3 melanoma — The pivotal randomized readout the whole category hangs on
- ClinicalTrials.gov — autogene cevumeran IMCODE-003 (NCT05968326) — BioNTech/Genentech's randomized phase 2 in pancreatic cancer
The hardest part is manufacturing
For an individualized vaccine, the chemistry-manufacturing-and-controls (CMC) section is where the regulatory difficulty concentrates. The product is synthesized fresh for each patient on a clinically actionable "vein-to-vein" timeline of days to weeks — sequence the tumor, design the construct, transcribe the mRNA, encapsulate it in a lipid nanoparticle, run release testing, and dose — with no opportunity to fully characterize a fixed finished batch the way a conventional biologic is. The regulator's leverage is to validate the process and the release specifications, then rely on comparability so a per-patient sequence variant does not trigger fresh full characterization each time.
The FDA has been signaling flexibility here. In early 2026 it publicized a "flexible approach" to CMC for cell and gene therapies — including not demanding overly onerous comparability data for minor manufacturing changes — which maps directly onto the per-patient-product problem. Manufacturing capacity and turnaround are themselves part of what regulators review: a platform that cannot reliably produce thousands of bespoke products on time is not approvable, however good the clinical data.
- FDA's "flexible approach" to CMC for cell & gene therapies (Covington, Jan 2026) — Accommodation for per-patient products and minor process changes
- FDA — Interactions with the Office of Therapeutic Products (CBER) — These are biologics: early engagement runs through CBER via INTERACT and pre-IND meetings
A different bar abroad
Western regulators are broadly aligned. Europe's PRIME scheme — the EMA's analog to Breakthrough Therapy — was granted to intismeran autogene for adjuvant melanoma in April 2023, again on promising-but-still-trial-gated terms. The divergence is elsewhere. Russia's Federal Medical-Biological Agency has cleared personalized neoantigen mRNA vaccines (Enteromix, then Oncorna) for clinical use largely on preclinical and early-phase data, without the large randomized confirmatory trials the FDA and EMA require.
For an informed reader the lesson is to weight approvals by the standard behind them. A clearance granted off preclinical results is not evidence a product would survive an FDA review, where a randomized phase 3 with a hard endpoint remains the gate. Regulatory divergence reflects the evidentiary bar, not necessarily the merit of the product.
- Merck — EMA PRIME designation for mRNA-4157/V940 (Apr 6 2023) — Europe's equivalent early-support designation
- Russia's FMBA approves a second personalized cancer vaccine, Oncorna (GxP News, 2026) — Clearance on early data — a markedly lower evidentiary bar
The open questions
A first approval would do more than clear one product — it would bless the whole "process is the product" model for biologics, establishing how a single marketing application can cover an open-ended family of per-patient molecules. Several things stay unsettled until then. How the FDA polices lot-to-lot comparability when every lot is a unique sequence is still being worked out; its 2026 flexible posture suggests accommodation but leaves the exact release-specification and post-approval-change rules open. Reimbursement of an individualized biologic — how a payer values a platform rather than a fixed product code — sits downstream of the FDA but is a real gating risk for the commercial story. And continuity of review for novel modalities has itself become a variable amid turnover among senior biologics reviewers.
The throughline: the science of picking neoantigens is racing ahead, but whether these vaccines become products is now largely a regulatory and manufacturing question — one the field will answer, or not, with a handful of randomized phase 3 readouts over the next year or two. For the investor framing of that same set of catalysts, see the growth view; for what's moving week to week, start with today's brief.
- A computational blueprint for personalized cancer vaccines — The other half of the story: how the vaccine is designed, phase by phase
- Growth — how the field is being weighed — The investor view of the same pivotal readouts