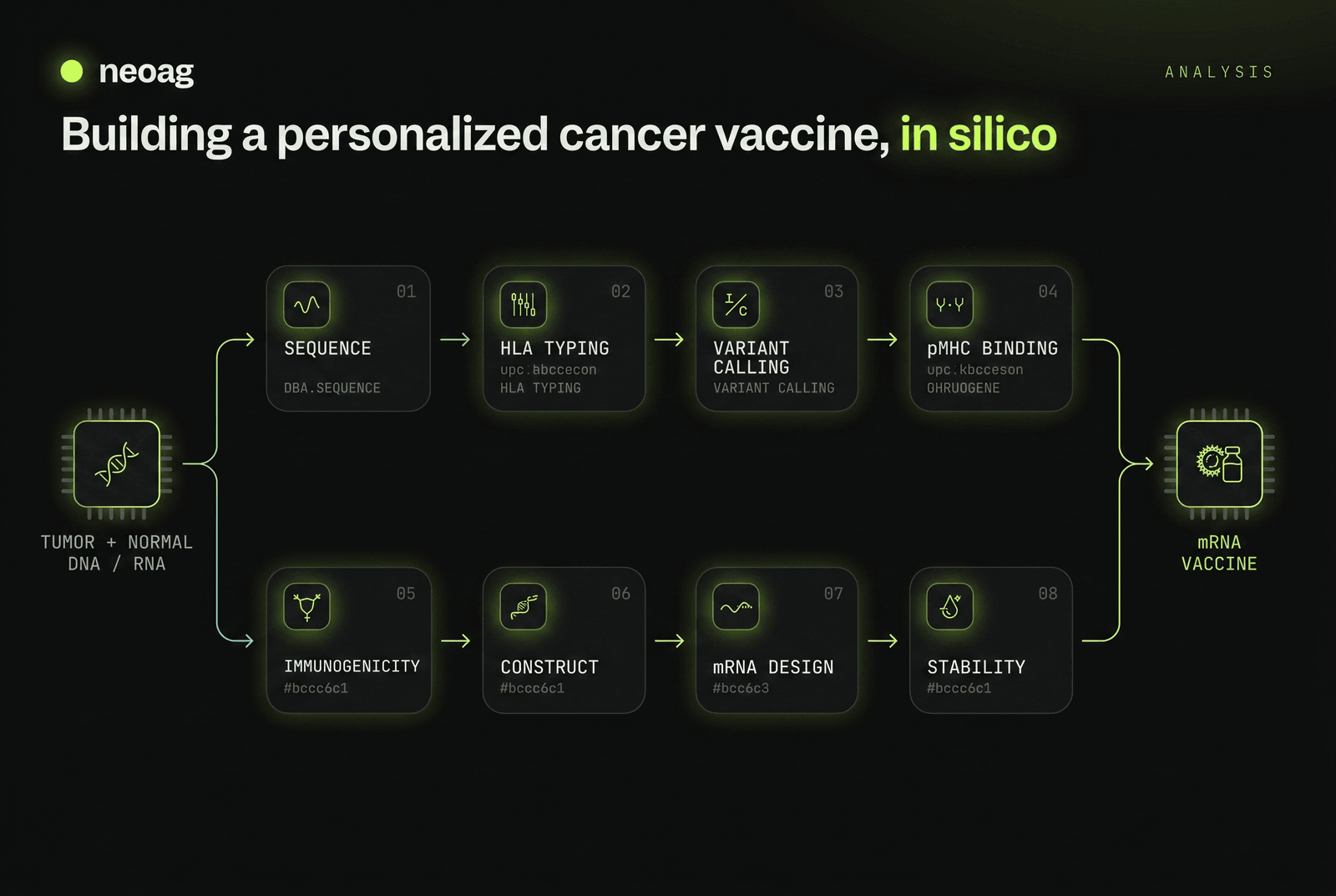
A personalized cancer vaccine is, at bottom, a long chain of computation. High-throughput sequencing and deep learning have together turned neoantigen discovery from artisanal guesswork into an industrial pipeline — one that ingests a patient's raw tumor and normal sequencing data and emits, at the far end, a stabilized mRNA construct ready for synthesis. Unlike shared tumor-associated antigens, neoantigens arise from mutations unique to one patient's tumor and absent from healthy tissue, which makes them precise targets — and makes every step of finding them a bespoke, in-silico problem.
This is a practical map of that pipeline: eight sequential phases, the specific open-source tools that run each one, the filtering thresholds that separate signal from noise, and the protein language models — many now hosted on Hugging Face — that define the current state of the art. It is the computational architecture only; the regulatory and GMP scaffolding around an investigational therapy — how the FDA approves a medicine that is different for every patient — is its own story, covered in the companion regulation piece linked at the end.
Nothing happens without sequencing. The pipeline needs three inputs in FASTQ form: tumor DNA (whole-exome or whole-genome) to capture the full landscape of alterations, a matched normal sample (usually blood) as a germline reference, and tumor RNA-seq to confirm that mutated genes are actually transcribed — a mutation that is never expressed can never be presented.
Reads are aligned to GRCh38 with BWA-MEM, sorted and indexed into BAM files. Because every variant caller has its own error profile, state-of-the-art pipelines run an ensemble — MuTect for low-allele-fraction variants, Strelka for SNVs and small indels — and take the union or intersection for a high-confidence call set. Tools like pVACfuse and AGFusion catch neoantigens from gene fusions and structural variants. Calls are written to VCF and annotated with the Variant Effect Predictor (VEP) to translate each DNA change into its altered amino-acid sequence. Finally, vt decompose splits multiallelic records, bam-readcount computes tumor variant allele frequency (VAF), and STAR + RSEM quantify expression as transcripts-per-million (TPM) — two numbers that drive prioritization later.
- BWA — Burrows-Wheeler Aligner; the BWA-MEM algorithm maps short reads to the reference genome.
- Strelka2 — Illumina; somatic SNV and small-indel caller, paired with MuTect in an ensemble.
- Ensembl VEP — Variant Effect Predictor; annotates how each DNA variant changes the encoded protein (run with --symbol --hgvs --canonical).
- STAR + RSEM — Splice-aware RNA-seq alignment (STAR) and expression quantification (RSEM) for per-transcript TPM.
- VAtools — Griffith Lab; utilities (incl. vcf-readcount-annotator) for folding VAF and expression back into the VCF for pVACtools.
For a peptide to be seen, it must be displayed by the patient's MHC molecules — encoded by the HLA complex, the single most polymorphic region of the human genome, with over 30,000 documented alleles. Each allele's binding groove admits a different repertoire of peptides, so the patient's exact six-allele Class I type (HLA-A, -B, -C) is a hard prerequisite for everything downstream.
OptiType is the field standard for typing directly from NGS reads. Ordinary mapping fails at the HLA locus because the alleles are too similar — reads multi-map everywhere. OptiType instead casts allele selection as an integer linear programming problem: it aligns reads against a library of all known alleles, then uses an ILP solver to find the diploid combination that explains the most reads while penalizing reads that map equally well to many alleles. Against 1000 Genomes truth calls it reaches near-perfect concordance, and a --beta parameter guards against falsely calling two alleles where the patient is homozygous.
- OptiType — ILP-based HLA Class I typing from NGS reads; runs RazerS3 mapping then a GLPK/CBC/CPLEX solver. `optitype run -i r1.fq -i r2.fq --dna -o results/`.
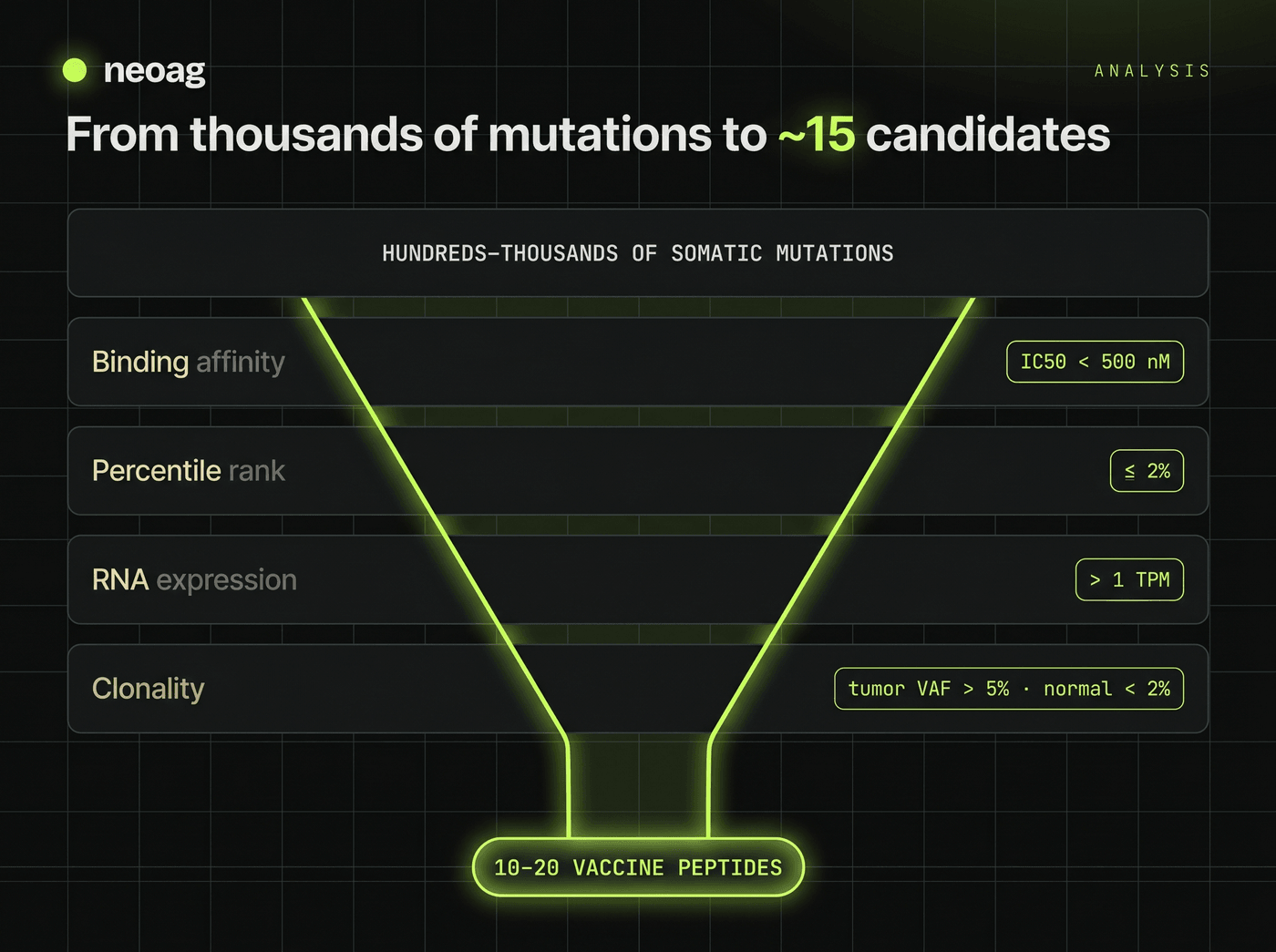
A tumor can carry hundreds to thousands of non-synonymous mutations. The job of this phase is to intersect those variants with the patient's HLA type, predict which mutated peptides bind, and filter aggressively. Two open-source ecosystems dominate. OpenVax — from Mount Sinai, and the computational backbone of trials like PGV-001 — is a modular Python suite: Vaxrank ranks candidate peptides on a composite of predicted Class I binding and variant abundance, Topiary handles the RNA-free fallback, and supporting libraries (PyEnsembl, Varcode, Isovar) translate variants into mutant protein sequences. pVACtools, from the McDonnell Genome Institute, is the most comprehensive and actively maintained suite, with a specialized module for each class of alteration.
| pVACtools module | Primary function |
|---|---|
| pVACseq | The primary engine — neoantigens from somatic SNVs and small indels. |
| pVACfuse | Neoantigens from structural variants and gene fusions (novel ORFs). |
| pVACsplice | Neoantigens from tumor-specific alternative splicing and exon skipping. |
| pVACbind | Binding affinity for raw, unannotated FASTA peptides against given alleles. |
| pVACview | Interactive R Shiny app for exploring and sorting the aggregated reports. |
The reason filtering is non-negotiable: up to 70% of initial in-silico predictions are false positives — peptides that won't bind in vivo, won't survive proteasomal cleavage, or won't provoke a T cell. pVACseq applies layered binding and coverage filters to distil thousands of candidates down to a workable 10–20, against thresholds grounded in tumor immunology.
- pVACtools — Griffith Lab; the comprehensive neoantigen suite. `pvacseq run <vcf> <sample> <alleles> <algorithms> <out>`, then binding_filter / coverage_filter.
- Vaxrank — OpenVax; ranks candidate peptides on combined Class I binding and variant abundance.
- Topiary — OpenVax; predicts mutated peptide-MHC ligands when RNA-seq evidence is missing.
- MHCflurry 2.0 — OpenVax; open-source pan-allele MHC-I presentation predictor, callable as a pVACseq backend.
| Metric | Threshold | Rationale |
|---|---|---|
| Binding affinity (IC50) | < 500 nM | Enough biochemical affinity to anchor in the MHC groove; strong binders are often < 50 nM. |
| Percentile rank | ≤ 2% | Affinity relative to a random-peptide background; normalizes across diverse alleles (≤ 0.5% = strong). |
| RNA expression | > 1–5 TPM | Confirms the mutated allele is actively transcribed — non-expressed mutations can't be presented. |
| Tumor DNA VAF | > 5% | Ensures a clonal fraction; subclonal mutations leave part of the tumor untargeted. |
| Normal DNA VAF | < 2% | Confirms the variant is somatic — a germline target risks lethal autoimmune toxicity. |
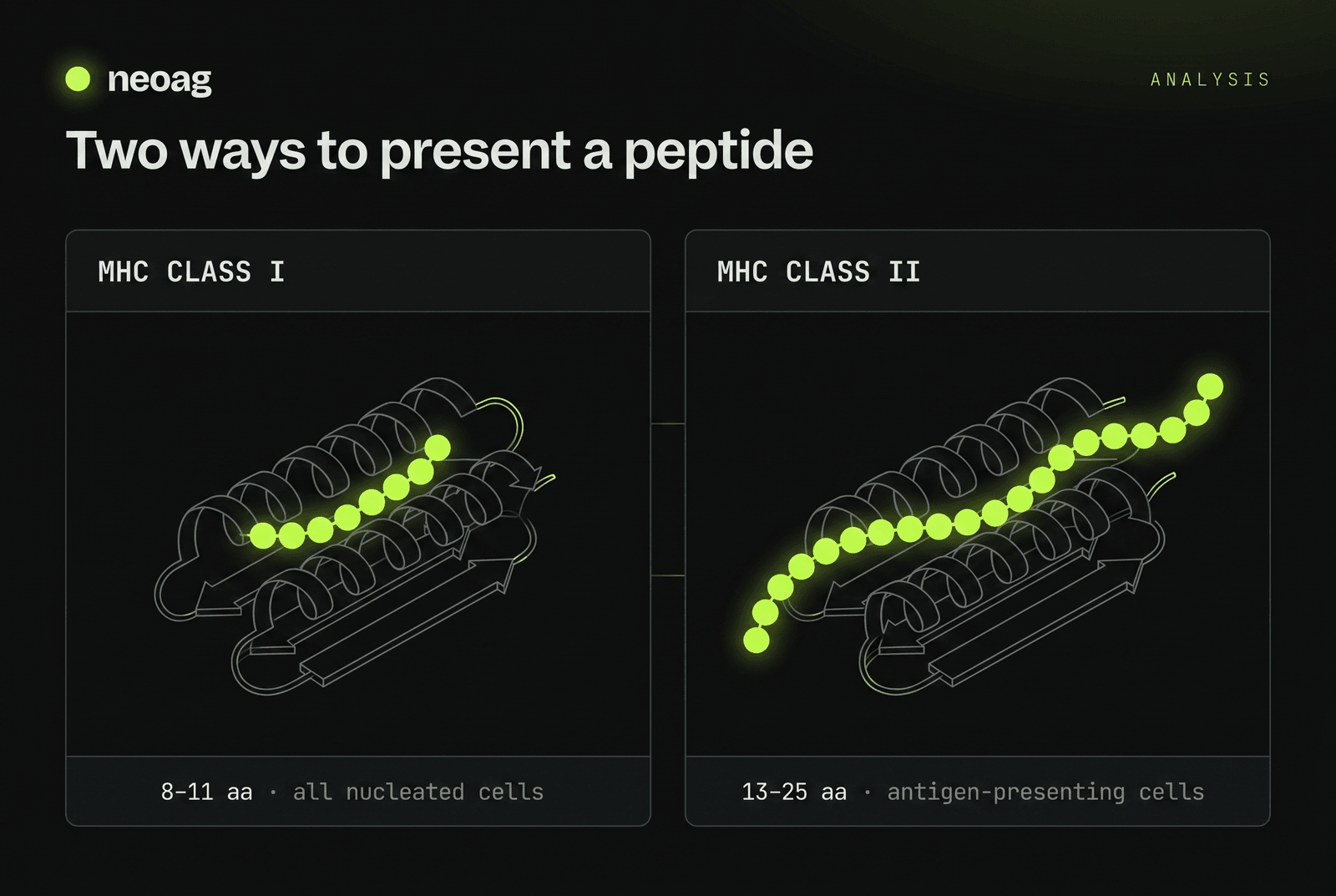
For a decade NetMHCpan — a shallow network — was the gold standard for peptide-MHC binding, but it falters on rare alleles and unusual peptides where training data is thin. The field has since reframed the problem in the language of NLP: a protein sequence has a grammar, and transformer-based protein language models (pLMs) can learn it. The biophysics they have to capture is sharp. MHC Class I, on every nucleated cell, has a closed groove that holds short 8–11-mers (9-mers dominate). MHC Class II, on professional antigen-presenting cells, has an open-ended groove that admits long 13–25-mers — far harder to predict, because the peptide register shifts and high-quality training data is scarce.
The pivot point is ESM-2 (Evolutionary Scale Modeling) from Meta AI — trained with masked-language modeling on 250M+ protein sequences, it generates context-aware embeddings from a single sequence with no expensive multiple-sequence alignment at inference. Because the base model knows general protein biology but not immunology, researchers adapt it with LoRA — a parameter-efficient fine-tune that freezes the base weights and trains small rank-decomposition matrices inside the attention layers. Hugging Face has become the registry for the resulting specialized models, deployable with a few lines of the transformers library.
- ESM-2 (650M) — Meta AI; the protein language model that underpins most of the models below — the fine-tuning base layer.
- smares/ESMCBA — Continued ESM pre-training fine-tuned on IEDB functional assays for MHC-I binding; median Spearman 0.62 across 25 alleles, beating NetMHCpan (0.56) and MHCflurry (0.49), and strong on low-resource alleles.
- O047/esm2_MHC-II_Reforged_Single — 650M ESM-2 + LoRA (r=16, α=32) with a dual BA/EL head; trained on ~1.5M eluted ligands across 200 HLA-II alleles. Input bundles peptide + 34-residue pseudosequence + flanking context for the open groove.
- ardigen/ardisplay-i — Ardigen; models the full HLA-I presentation pathway (TAP, processing, degradation), not just binding — >2× the Average Precision of standard tools on colorectal/glioblastoma cohorts. Peptides must be 8–11 aa.
- ChatterjeeLab/PeptiVerse — Cross-attention transformer pairing pooled embeddings with support-vector regression for peptide-MHC binding.
Presentation is not recognition. A peptide can bind beautifully and still draw no response, because thymic selection deletes T cells that react to self — and since neoantigens are often single-residue variants of self-peptides, the matching T cells may simply not exist. Predicting whether a T-cell receptor will actually engage a peptide-MHC complex is the field's hardest open problem, the "holy grail" of systems immunology.
The response has been to train on validated immunogenicity rather than mere binding. Seq2Neo and CNNeo bundle custom CNNs trained on epitopes with confirmed T-cell responses; NeoTImmuML draws on the upgraded TumorAgDB2.0 and runs a weighted ensemble of LightGBM, XGBoost and Random Forest over peptide physicochemistry. To model the physical TCR-pMHC interface directly, transformer models have entered the Hugging Face ecosystem: DecoderTCR continually pre-trains an ESM-2 backbone on MHC ligandomes and VDJdb interaction data to score binding events, while BERTrand attacks the field's chronic failure mode — collapse on unseen peptides — reaching 0.66 cross-peptide AUROC and showing that attention can learn the grammar of TCR engagement for genuinely novel neoantigens.
- biohub/DecoderTCR — ESM-2 backbone with continual pre-training on MHC Motif Atlas ligandomes + VDJdb; scores TCR-pMHC interface energy and binding.
With 10–20 immunogenic peptides chosen, the work turns from discovery to engineering. Concatenating short peptides into one mRNA or DNA vector produces a single polyprotein — and the seams between peptides create artificial junctional epitopes. If a junction happens to bind the patient's HLA, the immune system attacks the seam instead of the tumor, diluting efficacy and risking off-target toxicity.
pVACvector exists to defeat this. It scores every possible ordering of the peptides and tests non-immunogenic spacer sequences between them. Because the combinatorial space is astronomical, it explores with simulated annealing — accepting temporary worse arrangements to escape local minima — and converges on an ordering that minimizes junctional immunogenicity. If no spacer can rescue a junction, it clips leading or trailing residues (up to --max-clip-length) to dismantle the offending binding core while leaving each neoantigen payload intact.
| Spacer | Function |
|---|---|
| AAY, AAL | Standard flexible linkers from early multi-epitope constructs. |
| HHHH, HHAA, HHHC | Histidine-rich linkers that optimize proteasomal cleavage. |
| GGS, GPGPG | Glycine/proline-rich linkers that disrupt secondary structure, keeping the polyprotein cleavable. |
The polyprotein must now be reverse-translated to nucleotides — and because the genetic code is redundant, one protein maps to astronomically many mRNA sequences. The choice of codons and the resulting secondary structure govern shelf-life, translation rate, and how much antigen actually reaches the immune system. VaxPress (and its web counterpart VaxLab) orchestrates this: from a coding-sequence FASTA it iteratively optimizes for stability and expression, scoring designs on Codon Adaptation Index, GC content, repeat frequency, and minimum free energy via ViennaRNA, with a pluggable Python interface for custom fitness functions.
Two deep-learning advances sharpen the search. CodonBERT (from Sanofi) abandons frequency-based codon swapping for a transformer that reads whole codon triplets — trained with masked-language modeling on 10M+ mRNA sequences, it learns the contextual codon-pair rules that pace the ribosome and shape co-translational folding. LinearDesign, meanwhile, attacks structure: classical RNA folding scales at O(n³), prohibitive for long constructs, so it borrows from computational linguistics — a 5′-to-3′ dynamic-programming pass with beam search over a "codon graph" — to reach linear O(n) time and emit the sequence with the lowest deterministic free energy, with CAI folded into the edge weights.
- VaxPress / VaxLab — Chang Lab (SNU); CLI + web optimizer scoring CAI, GC, repeats and MFE (ViennaRNA), with pluggable fitness functions.
- CodonBERT — Sanofi; codon-triplet transformer trained on 10M+ mRNAs, learning contextual codon rules that legacy frequency methods miss.
- LinearDesign — Linear-time mRNA design via a codon-graph dynamic program + beam search; minimizes free energy while optimizing CAI.
The last check is whether the designed mRNA will survive. In-line hydrolysis — backbone cleavage driven by water and local structural strain — is the single biggest threat to mRNA-vaccine storage and distribution, so degradation is modeled before the expensive step of physical synthesis. DegScore, a ridge-regression model built on nucleotide-resolution degradation maps from the Eterna citizen-science platform, scores any sequence's vulnerability; RNAdegformer goes further with a neural architecture that maps degradation hotspots across full vaccine-length sequences, outperforming DegScore and simpler unpaired-probability metrics.
Chemical stability in the vial is only half the problem. In-vivo half-life is governed by codon optimality — transcripts rich in "optimal" codons resist deadenylation and decay. iCodon, an R package and web app, predicts species-specific mRNA stability from codon composition alone and uses an evolutionary algorithm to tune it in either direction: maximally stabilized for prolonged antigen expression, or deliberately destabilized for a transient, controlled release profile.
What the eight phases have in common is that biological intuition has been replaced by mathematics at scale: integer linear programming for HLA typing, ensemble variant calling, layered immunology filters, protein language models for binding and immunogenicity, simulated annealing for construct order, and linear-time dynamic programming for mRNA structure. The accuracy gains increasingly come from the handoffs between stages as much as from any single model — and from the shift to transformers that capture deep evolutionary context where shallow regression matrices could not.
Two cautions before treating any of these tools as plug-and-play. "Open" is not the same as "licensed for your use" — several models are non-commercial only, which matters the moment a program turns clinical. And no single tool is the pipeline; real neoantigen discovery chains all eight phases together. If the terminology here is unfamiliar, the primer and glossary cover the underlying biology; for the open models you can actually fine-tune at the prediction steps, see the companion analysis; and for what's moving week to week across exactly these methods, start with today's brief.
- How the FDA regulates an N-of-1 cancer vaccine — The other half of the story: how a medicine that's different for every patient is allowed to reach them